Moving one step beyond to explain the neurobiology of this global epidemic

Introduction
Depression, a global epidemic, is a leading cause of disability worldwide. According to WHO, globally more than 264 million people of all ages suffer from depression most of which can lead to suicide. More women are affected by depression than men. Back from the mid 20th century till now, several psychopharmacological drugs have been discovered to combat this disease all of whose mechanism of action remains overall the same. But currently, questions and debates have started to arise regarding its validity and efficacy in treating this disease.
So to date were our understanding regarding it lacking a strong foundation?
This question has sparked new research about a more detailed picture of the neurobiological basis behind this disease. But before going to that let me review a brief history of the development of antidepressants.
1.The Serendipitous Discovery of the first Antidepressant
It all started when in 1951, biochemists working at the Hoffman-La Roche laboratories in New Jersey were synthesizing hundreds of compounds to identify novel antimicrobial agents. Certain derivatives of hydrazine were discovered to be effective against Mycobacterium tuberculosis. One of the derivatives, known as iproniazid was successful in treating TB infection in humans. But some unusual and unexpected scenes occurred in the meantime. Patients treated with this new experimental drug reported some results which scientists thought side effects such as sunnier moods, increase in pleasure and appetite, improved sleep.
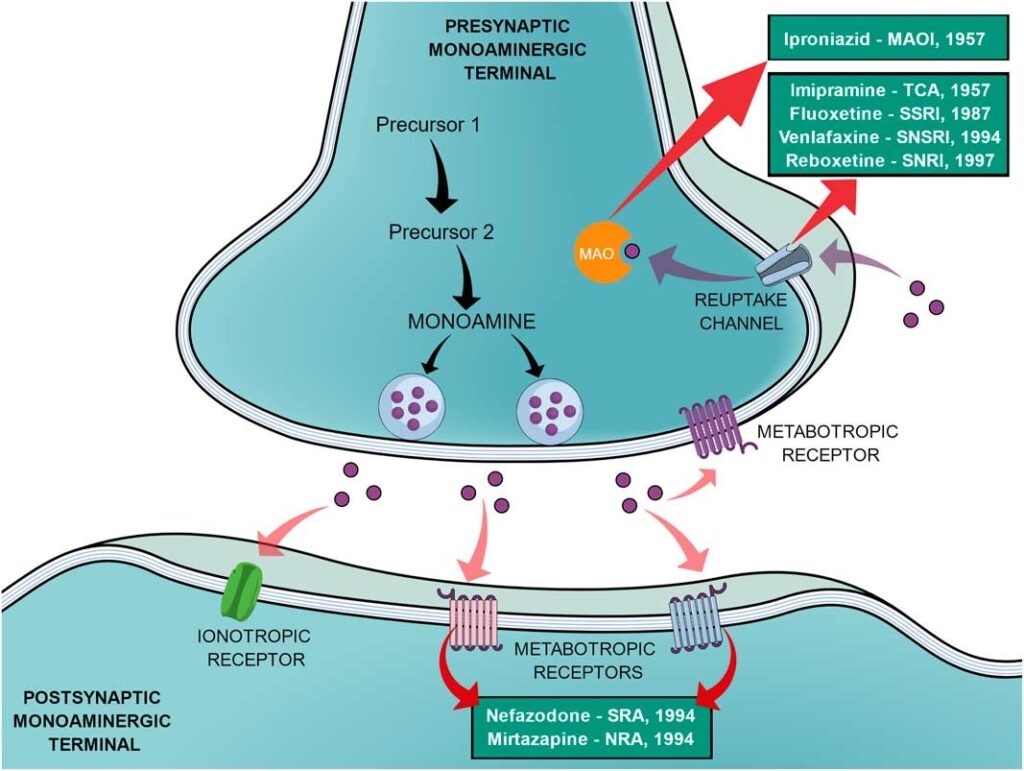
Soon after a lot of research, finally, in 1957, the novel TB drug iproniazid became the world’s first antidepressant drug. Later, it was found out that iproniazid works by inhibiting the enzyme, monoamine oxidase which breaks down monoamine neurotransmitters like serotonin, dopamine and noradrenaline which are involved in pleasurable emotions. Thus the first generation of antidepressant drugs came into existence called “Monoamine Oxidase Inhibitors”.

2.Emergence of Imipramine
Shortly after World War II, P. Charpentier and his colleagues in France synthesized and tested a series of phenothiazine amines, which was found to have significant and long-lasting antihistaminic properties. One of the most potent phenothiazine amines was promethazine. The therapeutic and commercial success of promethazine as a sedative and possible antipsychotic prompted the synthesis of many modified phenothiazines for their potential central nervous system (CNS) effects. Finally, the first antipsychotic, chlorpromazine, was synthesized when chlorine was added to the promethazine structure.
In 1945, Häfliger and Schinder, replaced the Sulphur bridge of the phenothiazine ring of promethazine with an ethylene bridge to synthesize G22355, a weak antihistamine and mild anticholinergic with sedative properties, later named imipramine. Thus, the first clinically useful Tricyclic Antidepressant (TCA) was discovered. Later, it was discovered that TCAs block the reuptake of noradrenaline.
Thus, the catecholamine hypothesis of depression came into existence
“Depression is caused by low levels of epinephrine and norepinephrine in the CNS”
3.Entry of Serotonin in the picture
At the same time, other findings contributed to the introduction of a new protagonist, 5- hydroxytryptamine (5-HT or serotonin) in the search for the molecule behind depression. In addition to the noradrenergic system, imipramine and iproniazide also affect the 5-HT system, and 5-HT neurotransmission seems to have an essential role in animal sedation. Later it was proposed that serotonin was a more important neurotransmitter in depression than noradrenaline.
Thus, the serotonergic theory of depression was postulated!
“Deficit of serotonin at synaptic cleft in certain brain regions lead to depression”
4.The rise of Prozac (fluoxetine)
Despite being the only alternatives for treating depression, the journey of iproniazid and imipramine soon came to a halt. The hepatotoxicity caused by iproniazid and symptoms like hypertension, drowsiness, vomiting and epigastric problems caused by iproniazid and imipramine slowly caused these classes of drugs to become obsolete. The need for more specific drugs with lesser side effects began to increase in the meantime. Thus for the first time in history, the postulation of the monoaminergic theories of depression led to a new search methodology for new therapeutic drugs.
The SSRIs(Selective Serotonin Reuptake Inhibitors) were the first class of drugs developed after a planned strategy that act in a specific site (the protein responsible for the serotonin reuptake, 5-HT). This strategy made it possible to avoid unspecific actions and culminated in more selective drugs and, consequently, decreased the frequency and probability of undesired effects. Subsequently, the pharmaceutical company Eli Lily created a ‘serotonin-depression study team’, composed by the researchers whose search for molecules that could selectively inhibit the reuptake of serotonin aimed to minimize collateral effects, such as cardiovascular toxicity and anticholinergic properties of the TCAs.
On July 24th, 1972, fluoxetine hydrochloride was designated the most powerful and selective inhibitor of serotonin uptake among all the compounds developed. In December 1987, the Food and Drug Administration (FDA) approved its clinical use, allowing Prozac to be released onto the market. By 1990, it was the most widely prescribed drug in North America and, in 1994, it was the second biggest selling drug in the world. After SSRIs, lots of other drugs based on overall same mechanisms came into existence like SNRIs(Serotonin-Norepinephrine Reuptake Inhibitors), NRIs(Norepinephrine Reuptake Inhibitors), NDRIs(Norepinephrine-Dopamine Reuptake Inhibitors).
But not all expectations about these drugs were fulfilled and instead several questions or confusions started to arise.
Question/Confusion 1: If monoamine levels(serotonin and norepinephrine) in synaptic cleft show an acute increase within few hours of taking SSRIs, why it takes 2 to 4 weeks for these drugs to show antidepressant effect?
Question/Confusion 2: Deficit of serotonin occurs in depressed people but on the contrary the decrease of serotonin levels in the brain caused by the acute depletion of tryptophan, serotonin’s precursor, does not induce a depressive-like behaviour in healthy humans.
Question/Confusion 3: Why SSRIs are only effective in appx 50-60% people while others remain unaltered?
All these however show that much more than monoaminergic neurotransmitter levels should be targeted in the brain of depressed individuals and much more studies are required for explaining the complex neurobiology of this disease. Let’s focus on some recent studies which give much clearer pictures about this disease.
Melanocortin – The Appetite Hormone
Stanford University School of Medicine scientists have laid bare a novel molecular mechanism responsible for the most important symptom of major depression: anhedonia, the loss of the ability to experience pleasure. While their study was conducted in mice, the brain circuit involved in this newly elucidated pathway is largely identical between rodents and humans. The study reveals how a hormone known to affect appetite turns off the brain’s ability to experience pleasure when an animal is stressed. This hormone, melanocortin, signals to an ancient and almost universal apparatus deep in the brain called the reward circuit, which has evolved to guide animals toward resources, behaviors and environments — such as food, sex and warmth — that enhance their prospects for survival.
A few scattered studies had suggested that chronic stress increased melanocortin levels in the brain. Stressed animals have heightened numbers of receptors for melanocortin in the nucleus accumbens, a key region of the reward circuit. If this all-important circuit is working properly, it doles out pleasure when an animal achieves a desirable goal or experiences rewarding stimuli, such as food or sex. If it’s not working properly, anhedonia is the result.
The scientists found that both chronic stress and the direct administration of melanocortin diminished the signaling strength of some of the tiny electrochemical contacts, known as synapses, on a set of nerve cells in the nucleus accumbens that contain receptors for melanocortin.
The melanocortin pathway is already of interest to drug companies. So companies already have melanocortin mimics and inhibitors at their disposal that could be used in clinical tests to determine whether managing patients’ melanocortin signaling relieves anhedonia.
Ketamine – The Current Game Changer in Antidepressant Research
This drug started its journey as an anesthetic in the Vietnam War, but currently it is revolutionizing the field of antidepressant research. Ketamine not only works fast but also works in people who don’t respond to other antidepressants. About one-third of people with depression today have treatment-resistant depression (TRD). The excitement over ketamine among depression researchers and clinicians is clear. They point out that ketamine is the first drug with a new mechanism to treat depression in more than half a century. In March 2019, the US Food and Drug Administration showed just how much expectations have changed by approving Janssen Pharmaceuticals’ nasal spray containing esketamine, the S enantiomer of ketamine, as the first form of ketamine to treat depression, specifically TRD. (The S form of the drug binds to ketamine’s target in the brain more strongly than the R form.)
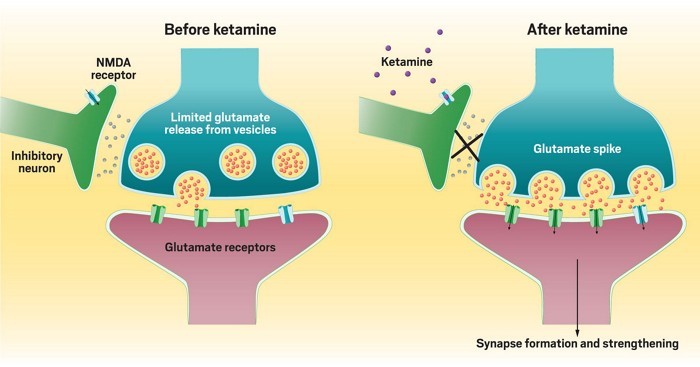
It all started back in late 1990s when researchers looked beyond the monoamines and focused on another neurotransmitter: glutamate. As the main excitatory neurotransmitter in the brain, it pushes neurons to fire electrically. Research at the time indicated that glutamate somehow regulated brain circuits implicated in depression. So they decided to test ketamine in part because the anesthetic blocks a glutamate receptor in the brain called the N-methyl-d-aspartate (NMDA) receptor. They observed patients unexpectedly feeling better within hours after a dose. But it is also associated with some serious side effects like elevated blood pressure and heart rate, psychological side effects—like anxiety and out-of-body experiences.
All these downsides to ketamine have led researchers to look for other molecules with rapid antidepressant effects but with less baggage. But unfortunately, a complete picture of how ketamine produces its antidepressant effect doesn’t exist, making it hard to know which targets in the brain are the best to go after.
Some scientists have looked at ketamine’s anesthetic target, the NMDA receptor. This receptor is an ion channel that allows calcium and sodium ions to flow into a neuron when opened by a combination of the right electrical conditions and the binding of glutamate and another molecule, sometimes glycine. Ketamine blocks this ion flow by binding inside the ion channel, essentially acting like a plug in a bathtub.
But the question is, How does ketamine trigger glutamate signaling by blocking the NMDA receptor? In the brain, some neurons tell other neurons to fire, while others tell those same neurons to stay quiet. Researchers think that ketamine blocks NMDA receptors on the neurons transmitting that quiet signal. Ketamine specifically blocks the NMDA receptor on inhibitory neurons and by blocking that, it is actually increasing activation. In other words, ketamine stops the inhibitory neurons from putting the brakes on, leading to an increase in glutamate signaling. But how ketamine could selectively target inhibitory neurons is not yet clear. But still it is really a textbook changer in this field which has narrowed and focussed research in this domain.
So finally back from the 1950s till now, the neurobiology of this devastating disease is still attracting research close to it. Some unexpected incidents had occurred in the history of antidepressant drugs. Some are partly successful, while some have failed in the long run. But the signs are promising with the emergence of ketamine and I am sure that one day will come when the word depression will be wiped out from the dictionary.
Till then, thank you and take care of your physical as well as your mental heath.
References:
- Pereira, V., & Hiroaki-Sato, V. (2018). A brief history of antidepressant drug development: From tricyclics to beyond ketamine. Acta Neuropsychiatrica, 30(6), 307-322. doi:10.1017/neu.2017.39
- Development and discovery of SSRI drugs (Wikipedia)
- Torrice, M (2020). Ketamine is revolutionizing antidepressant research, but we still don’t know how it works. ACS Neuroscience, 98(3).
About the author:

My name is SHRAMAN JANA. I am a 1st-year BS-MS undergrad in IISER Kolkata, KVPY fellow and recipient of NEET 2020 AIR 332. Being an aspired researcher by mind, I am a guy who has great affection towards biology and want to solve unknown and unanswered stuff in this subject. Apart from studies, I love reading storybooks, singing, play and watch football and cricket, play computer games and a die-hard fan of FC Barcelona. I always believe in sharing knowledge and follows the quote- ”If you want to shine like a sun, first burn like a sun.”
This is the first blog of Shraman for The Qrius Rhino.
2 thoughts on “Is Monoamine Theory Sufficient To Explain Depression?”