
Designed by Diptatanu Das.
The Problem
The human genome contains approximately 3 billion nucleotide base pairs and every time a human cell divides, it must copy all of these base pairs with high fidelity. However, it has been estimated that DNA polymerases make errors approximately once every 104–105 nucleotides polymerized, which means, that there can be an unavoidable error of approximately tens of thousands to hundreds of thousands of base pairs of DNAs per cycle of cell division. Proofreading can enhance the overall fidelity of DNA synthesis by a factor of 102–103. (Bębenek & Ziuzia-Graczyk, 2018; Echols & Goodman, 1991; Kunkel, 2009)
Even then, there is always a fair probability of base pairing errors, especially in the genes that can promote cancer. The cell has several pathways to keep a check of these errors, including oncogene-mediated apoptosis (programmed cell death) and senescence (cell ageing). (Reimann et al., 2010) However, in the presence of certain factors (like, environmental stress, UV, etc.) or chemicals (like, carcinogens, etc.), there is an increase in the probability of base pairing errors which may ultimately introduce errors in the coding region of DNA protecting genes or tumour-suppressor gene, etc, causing disruption of the protective pathways. This can lead to an accumulation of cells with erroneous genes, especially the ones that lead to tumour or cancer. In such cases, the probability of failure of natural protection increases so much that tumorigenic cells develop. Once formed, these tumour cells can evade immune protection using two main strategies:
- avoiding immune recognition
- instigating an immunosuppressive signal
(Gonzalez et al., 2018)
The Paradox
Our understanding above suggests that, chances of developing cancer should increase with:
- increase in the number of cells in the organism
- higher surface area of cells exposed to factors or chemicals that promotes erroneous genetic duplication/transcription/translation
- higher exposure time to such factors or chemicals, or a higher lifetime of the organism
Therefore, it can be expected that larger the animal is, greater is its probability to develop cancer. However, interestingly, it was first observed by English statistician and epidemiologist Richard Peto that humans not only contain 1000 times more cells but also lives 30 times longer than mice, yet the two species do not suffer incredibly different probabilities of developing cancer, that is, our expected correlation of cancer with the body mass and lifetime of the organism, fails.
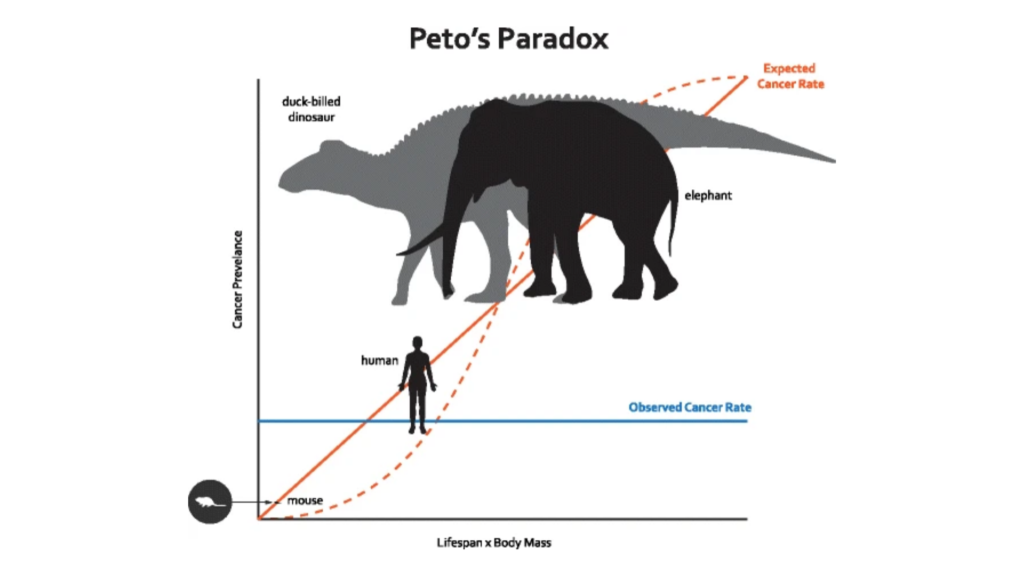
Reason?
The best guess that can explain this paradox is evolution. An organism that faces such high probabilities of developing a tumour must also evolve additional or more efficient protective strategies to adapt and survive. Indeed, it has been found that the genome of the African savannah elephant (Loxodonta africana) contains 20 copies of the tumour suppressor gene TP53. Now, this is an enhancer for the extra LIF6 gene which is a leukaemia inhibitory factor (LIF) that elephants have. TP53 is expressed in response to DNA damage, which, in turn, transcriptionally upregulates the activity of LIF6.
Now, what is very interesting is that LIF6 is a pseudogene. A pseudogene consists of one or more non-functional segments of DNA that resemble functional genes, which can form either by reverse transcription of functional mRNA transcripts, or simply (or, classically) by undergoing a mutation in one of its copies, that renders it non-functional. Normally, if a pseudogene undergoes re-functionalization, it has two issues or concerns:
- Needs a new cis-regulatory element (DNA segment that will promote binding of its transcriptional regulator or trans-acting regulatory protein)
- Successful re-functionalization might lead to toxicity over a long time (when the active gene is not required)
These problems can be solved by the presence of an inducible cis-acting element on the pseudogene, which can be switched on or off as per biological requirement. TP53 provides such a scope in the case of LIF6.
LIF6, on getting activated, promotes apoptosis of the cells by mitochondrial dysfunction, via the Bak/Bax-dependent pathway. Other pathways involving the activity of tumour suppressive genes are also reported, like the CDKN2A tumour suppressor pathway in naked mole-rats (Heterocephalus glaber).
Another interesting mechanism, which may explain this problem, is the ‘hypertumour’ hypothesis. This suggests that bigger tumours in bigger animals take a longer time to grow. This makes them susceptible to ‘cheater’ cells, which can take advantage of the tumour’s angiogenic (formation of new blood vessels from pre-existing vessels) properties in terms of nutrition and gaseous exchange. This creates a kind of competitive circumstance, which lowers the fitness of the whole tumour, thus, lowering the overall lethality of cancer in the bodies of large animals.
Some other adaptations in larger animals include:
- increased immunocompetence – better ‘attack’ on tumorigenic cells.
- shortened telomere – shorter protective terminal capping of chromosomes will cause reduction of the number of cycles the same cell can divide in an immortalized fashion.
(Tollis et al., 2017; Vazquez et al., 2018)
An Answer to Cancer Therapy?
As we now know some of the basic molecular schemes that justifies Peto’s paradox, we can understand that some of the mechanisms mentioned above can actually be used in treating tumours or cancers. The rationale can run the gamut from induced re-functionalization of genes (externally introduced or de novo hyper-activation if identified) to artificially playing with the tumour volume and frequency of ‘cheater’ cells. Immunotherapy is currently an actively pursued therapy which is under further improvement. Shortening of telomere might be a more complicated strategy if undertaken artificially, which might have severe consequences from collateral damages. Other naturally occurring strategies that explain Peto’s paradox should be actively hunted for. This can be considered as a silver lining to the challenges that we face in terms of developing safer and more effective targeted cancer therapy. Who knows, a gold standard answer to cancer therapy, which is still unknown to us, might actually lie in such naturally occurring mechanisms, which might even be the fruit of the discovery of other such paradoxical occurring.
By Diptatanu Das (Department of Biological Sciences, IISER Kolkata)
Edited by Diptomit Biswas (Department of Biological Sciences, IISER Kolkata)
References:
Bębenek, A., & Ziuzia-Graczyk, I. (2018). Fidelity of DNA replication—a matter of proofreading. Current Genetics, 64(5). https://doi.org/10.1007/s00294-018-0820-1
Echols, H., & Goodman, M. F. (1991). Fidelity Mechanisms in DNA Replication. Annual Review of Biochemistry, 60(1). https://doi.org/10.1146/annurev.bi.60.070191.002401
Gonzalez, H., Hagerling, C., & Werb, Z. (2018). Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes & Development, 32(19–20). https://doi.org/10.1101/gad.314617.118
Kunkel, T. A. (2009). Evolving Views of DNA Replication (In)Fidelity. Cold Spring Harbor Symposia on Quantitative Biology, 74(0). https://doi.org/10.1101/sqb.2009.74.027
Reimann, M., Lee, S., Loddenkemper, C., Dörr, J. R., Tabor, V., Aichele, P., Stein, H., Dörken, B., Jenuwein, T., & Schmitt, C. A. (2010). Tumor Stroma-Derived TGF-β Limits Myc-Driven Lymphomagenesis via Suv39h1-Dependent Senescence. Cancer Cell, 17(3). https://doi.org/10.1016/j.ccr.2009.12.043
Tollis, M., Boddy, A. M., & Maley, C. C. (2017). Peto’s Paradox: how has evolution solved the problem of cancer prevention? BMC Biology, 15(1). https://doi.org/10.1186/s12915-017-0401-7
Vazquez, J. M., Sulak, M., Chigurupati, S., & Lynch, V. J. (2018). A Zombie LIF Gene in Elephants Is Upregulated by TP53 to Induce Apoptosis in Response to DNA Damage. Cell Reports, 24(7). https://doi.org/10.1016/j.celrep.2018.07.042
Cover Picture designed by Diptatanu Das using elements from “Cancer cell cartoon PNG Designed By 天边 from https://pngtree.com” and Canva
About the editor:

Diptomit is a 5th-year BS-MS Student at IISER Kolkata and an INSPIRE Scholar, majoring in biology. He has been a part of the 2019 gold medallist iGEM IISER Kolkata team. Besides being addicted to PC games, he loves to spend his free time either in the gym or in bed thinking about philosophy and the meaning of human existence. His interest in pursuing science lies solely in venturing into the unknown and coming across patterns and observations which shed more light on the mysteries of nature.
This is his first contribution to help TQR initiative grow.
2 thoughts on “Peto’s Paradox – An Answer to Cancer Therapy?”